

Amy Llancari
My project focuses on differentiating the progression of Non-alcoholic Fatty Liver Disease (NALFD) into Hepatocellular Carcinoma (HCC) between male and female mice. HCC is the sixth most prevalent cancer in the world, yet it ranks as the third leading type of cancer deaths with an accelerated increase in incidence rates. NAFLD is a major risk factor for HCC, and is emerging as the most common chronic liver condition in the Western world. Prevalence of both HCC and NAFLD is significantly higher in men than women worldwide, suggesting sex-specific differences may play a role in disease susceptibility. My project will focus on investigating these sex-specific morphological and cell expression pattern differences. This will provide insight on potential protective mechanisms found in female mice inhibiting liver injury, in addition to potential diagnostic markers and molecular treatment targets during NAFLD progression.
I have been part of the Porter since my first year of undergrad. My time in the Porter Lab has been the highlight of my university career. I cannot put into words how thankful I am for the unwavering support and guidance I’ve received throughout my project. Dr. Porter and the RA’s have been exceptional role models, their dedication to the scientific community is truly inspiring, and their passion for research is contagious. Being a part of this incredible team has been an honor, and has provided me with so many opportunities both in research and community involvement. My time in the lab has taught me valuable research skills, as well as transferable skills that I will carry with me throughout my career. As I pursue my Masters in the Porter Lab, I am excited to continue to learn and grow as a researcher.
Alan Cieslukowski
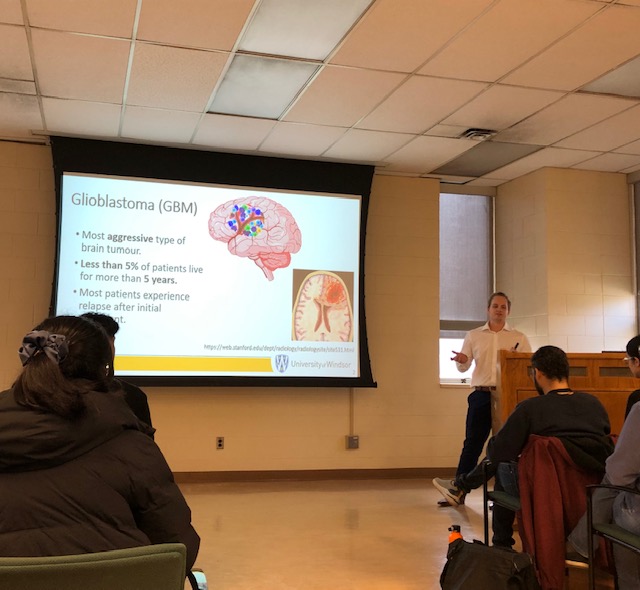
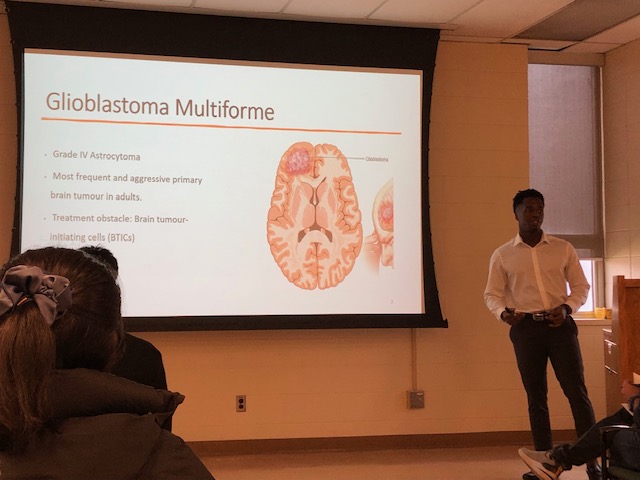
Efficient targeting of multiple components of a tumour might be a successful strategy in aggressive types of cancer such as glioblastoma (GBM), which remains the most common and malignant primary brain tumour with an extremely poor patient survival of less than 15 months. The significant therapeutic challenge posed by GBM stems from its genetic and phenotypic heterogeneity fueled by characteristics such as aggressive and treatment-resistant populations of Tumour Initiating Cells (TICs) and high levels of angiogenesis contributing to tumour evolution. TICs, which are responsible for GBM patient relapse, thrive in niches close to blood vessels where they interact with endothelial cells (ECs), exit the cell cycle, and evade therapies. Targeted antiangiogenic drugs, preventing GBM cells from recruiting new blood vessels are only temporarily effective in 50% of patients, resulting in acquired secondary resistance by the tumour. This project explores the TIC-EC interplay and its role in propagating tumour aggressiveness and therapy resistance. This project investigates the impact of ECs on the aggressive characteristics of individual, specific populations of TICs using GBM patient-derived systems, including 3D organoid models and zebrafish patient-derived xenografts (PDXs). Considering that NOTCH1 signaling regulates both TIC and EC populations in GBM, this project dissects the role of NOTCH pathway in TIC-EC interaction. Elucidating the details of cell cycle signaling and specific populations of aggressive TICs with dependence on the EC component will contribute to the identification of new and better therapeutic targets and personalized approaches to treatment of patients with GBM in the future.

Depen Sharma
Tuberous Sclerosis Complex (TSC) is a genetic disorder affecting 1 in 6,000 births, leading to benign tumour formation in various body parts such as the brain, kidney, skin, and lungs. This condition arises from mutations in the TSC1 or TSC2 genes and presents a wide range of neurological symptoms, making diagnosis challenging. Tuberin, a protein encoded by the TSC2 gene, is critical in regulating cell growth, proliferation, and the cell cycle. A key element in TSC is the interaction between Tuberin and Hamartin, encoded by the TSC1 gene. Hamartin ensures Tuberin stability by inhibiting its interaction with the E3-ligase HERC1, which would otherwise lead to Tuberin degradation. Tuberin also regulates the G1/S and G2/M checkpoints in the cell cycle, which are crucial for controlling cell growth and division.
Our research explores the potential role of Cyclin B1, a cell cycle protein, in stabilizing Tuberin levels without Hamartin. Cyclin B1 is essential for mitotic entry, and its function has yet to be entirely understood. It has five serine residues that can be mutated to create a phospho-null Cyclin B1 construct (Cyclin B1 5A), which we utilize in our investigation. Previous work has shown that Tuberin can bind Cyclin B1 during the G2 phase of the cell cycle, but more is needed to know about their interactions when Hamartin is present. Cyclin B1 may be essential in Tuberin stabilization during the G2/M transition. Understanding the relationship between Cyclin B1 and Tuberin in TSC could provide valuable insights into the disorder’s molecular mechanisms and potentially lead to novel therapeutic approaches. By elucidating the role of Cyclin B1 in Tuberin stabilization, we hope to contribute to the ongoing efforts in understanding and treating Tuberous Sclerosis Complex.
Emmanuel Boujeke

Lia Oschanney
Liver cancer is one of the leading causes of cancer death globally, of which 90% of cases are Hepatocellular Carcinoma (HCC). One risk factor of HCC is Non-Alcoholic Fatty Liver Disease (NAFLD), often triggered by sedentary lifestyle, poor diet, and obesity. Most cases of HCC are cirrhotic and can start from excess fat accumulation in NAFLD. Chronic fat can trigger inflammatory responses that damage cells, leading to the liver initiating a proliferative response to recover. However, over time the liver shifts to a fibrotic state to maintain structural integrity with low levels of proliferation, characteristic of Non-Alcoholic Steatohepatitis (NASH). As fibrosis becomes extensive, the liver becomes cirrhotic, and the environment increases HCC risk; however, 20% of HCC cases are non-cirrhotic arising directly from NAFLD. The atypical cyclin-like protein Spy1 is capable of binding to CDKs, increasing cell division and overriding checkpoints. Spy1 has been shown to have a role in cancers, including HCC. Previous research has shown that mice with an overexpression of Spy1 in the liver develop NAFLD and HCC with decreased fibrosis, suggesting that elevation of Spy1 drives a proliferative response in the presence of cell damage, thus increasing HCC risk. To better understand the mechanisms that govern this response and the role of Spy1, this project examines NAFLD development in a transgenic mouse model driving Spy1 overexpression in the liver. This work may lead to the use of Spy1 as a potential drug target in the treatment of HCC.
In my first year, I knew I had a strong interest in research, but I was unsure how to take the first steps towards getting involved. Then, I discovered Dr. Porter’s website and was immediately captivated by the amazing work being conducted in the lab. However, just as I was eager to get started, the pandemic hit, and I was unable to set foot in the lab for nearly two years. Despite this obstacle, the lab kept me engaged through peer mentoring, which continued to fuel my curiosity and teach me the fundamental principles underlying research. When I finally had the opportunity to work in the lab as a third-year student, I had limited practical experience due to the majority of my undergraduate studies occurring online. However, under the guidance of the Porter lab team, I learned a multitude of techniques and quickly progressed from a hesitant and uncertain beginner to a confident and independent researcher capable of working on my own project. This experience has not only prepared me for future research endeavors but has also equipped me with invaluable skills that will undoubtedly serve me well in my future career in medicine. Completing my undergraduate thesis was an incredibly rewarding experience, and it would not have been possible without the support of my peers and the guidance of Dr. Bre-Anne Fifield and Dr. Lisa Porter. I am grateful for the opportunity to have worked in such an inspiring environment and to have learned from some of the brightest minds in the field.
