Glioblastoma (GBM) is the most frequent and aggressive primary malignant brain tumour. Despite rigorous therapeutic efforts, the prognosis of GBM remains extremely low, with a median survival of approximately 14 months after diagnosis. Our lab studies a cyclin-like protein called Spy1 which has been found to be elevated in GBM. Spy1 overrides cell-cycle checkpoints and avoids conditions that would normally cause cell cycle arrest, leading to increased and uncontrolled cell proliferation. Our research has demonstrated Spy1’s involvement in regulating senescence, a prolonged state of cell cycle arrest, specifically within the context of GBM. We have shown that targeting Spy1 leads to an increase in the number of senescent cells, and importantly, these senescent populations can be effectively eliminated using senolytics, drugs that can target and eliminate senescent cells.
I joined the Porter Lab at the beginning of my first year of university as an outstanding scholar. I started as a lab volunteer and a junior student in the Peer Mentor Network (PMN) where I learned the basics of cancer research from more experienced students. Over time, I took on a more active role, identifying knowledge gaps, assisting with experiments, and collaborating with peers to address current research challenges. Working on my own project in my upper years allowed me to develop my critical thinking and problem-solving skills, and taught me the benefits of working in a collaborative environment. This experience has ignited a passion within me for research and lifelong learning, and I am truly grateful to have had the opportunity to pursue research under the guidance of Dr. Porter and the research associates.
My project looked at the functional consequences of Tuberin mutations, more specifically the A614D and C696Y missense mutations. Mutations in this protein result in a disease called Tuberous Sclerosis Complex (TSC). TSC causes benign tumour formation in various organs of the body including the brain, heart, eyes, and lungs among others. These point mutations result in Tuberin losing its regulatory function over protein synthesis and mitotic onset. I investigated the punctate perinuclear localization pattern that is seen with these point mutations by looking at the lysosome as a potential cause for this. We hypothesize that Tuberin mislocalization to the lysosome results in a loss of Tuberin’s regulatory role over mitotic onset. I also aimed to develop cells that express these Tuberin mutations at physiological levels so we can better understand the interactions of these Tuberin mutants at endogenous levels.
I joined Porter lab in my 4th year of undergrad to do a thesis and my experience in the lab was great, being able to work in the lab allowed me to gain valuable skills in not only critical thinking and problem solving but also oral and written communication, every member of the lab was very kind and generous with their guidance. The skills that I developed in the lab will be extremely beneficial in the future and I want to thank everyone in the lab for their continued support throughout the year.


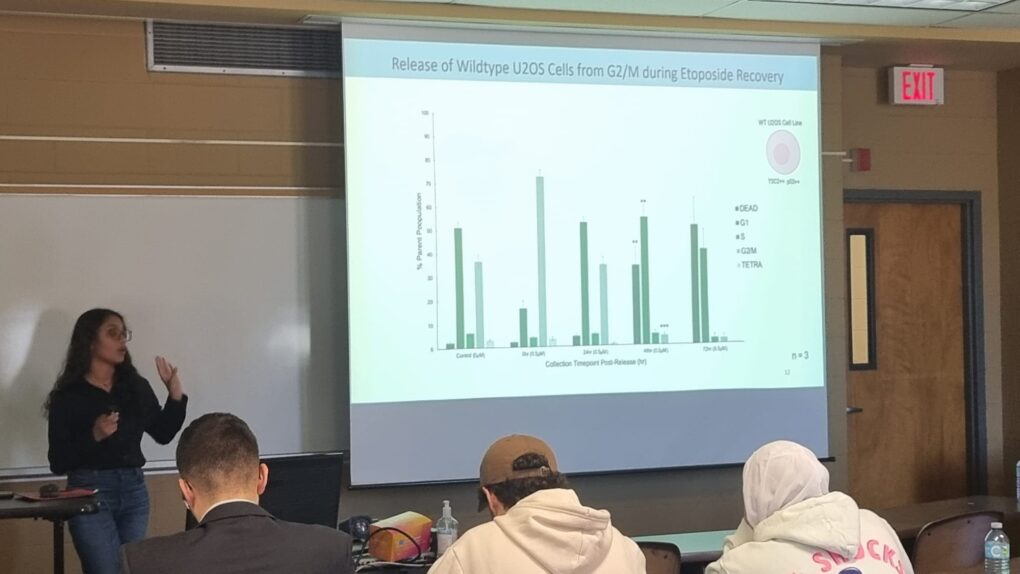

My project focuses on tool development and neuroendocrine prostate cancer. A standard treatment protocol for castrate-resistant prostate cancer involves the use of androgen receptor inhibition therapies. While effective, in some cases, androgen deprivation through treatment can pressure prostate cancer cells to differentiate into neuroendocrine prostate cancer cells (NEPC). Current treatment options for NEPC remain limited, and understanding the molecular mechanisms guiding this differentiation will be essential in developing future therapies. My research questions focuses on whether we can stop the cells from transforming into such an aggressive form of NEPC cells by using Cyclin-dependent kinase (CDK) inhibitors. I use LNCaP cell lines and the zebrafish animal model to test the hypothesis that elevated levels of Cyclin A and/or Cyclin B1 support the evolution to NEPC and that select targeting of their kinases, Cdk1 and/or Cdk2, at the G2M checkpoint, could represent an effective therapy for preventing progression and treating NEPC. Furthermore, I constructed fluorescent splitFAST vectors that allow real-time monitoring of the interaction between Cyclin B1 and Tuberin. Our lab has observed that Tuberin expression plays a role in embryonic neurodifferentiation and could be a potential player within NEPC differentiation. The fluorescent vectors allow us to visualize the effects of the CDK inhibitor. While research is constantly testing the unknown, there is strong rationale and data to support that this is a promising direction that could dramatically impact overall outcomes on a global scale.
I joined Porter Lab and my third year and it has been one of the most fulfilling experiences in my university career so far. I’m incredibly thankful to Dr. Porter and Ras, especially Dr. Elizabeth Fidalgo de Silva for supporting me and helping me in my journey as a scientist. I’m incredibly grateful for the Porter Lab community for supporting and being a great team. My time in Porter Lab has taught me invaluable skills that I am looking forward to carry on with me into my next steps for my science career.